Simplifying models of stem-cell dynamics in chronic myeloid leukemia
December 17, 2013 4 Comments
 If I had to identify one then my main allergy would be bloated models. Although I am happy to play with complicated insilications, if we are looking at heuristics where the exact physical basis of the model is not established then I prefer to build the simpleast possible model that is capable of producing the sort of results we need. In particular, I am often skeptical of agent based models because they are simple to build, but it is also deceptively easy to have the results depend on an arbitrary data-independent modeling decision — the curse of computing. Therefore, as soon as I saw the agent-based models for the effect of imatinib on stem-cells in chronic myeloid leukemia (Roeder et al., 2002; 2006; Horn et al., 2013 — the basic model is pictured above), I was overcome with the urge to replace it by a simpler system of differential equations.
If I had to identify one then my main allergy would be bloated models. Although I am happy to play with complicated insilications, if we are looking at heuristics where the exact physical basis of the model is not established then I prefer to build the simpleast possible model that is capable of producing the sort of results we need. In particular, I am often skeptical of agent based models because they are simple to build, but it is also deceptively easy to have the results depend on an arbitrary data-independent modeling decision — the curse of computing. Therefore, as soon as I saw the agent-based models for the effect of imatinib on stem-cells in chronic myeloid leukemia (Roeder et al., 2002; 2006; Horn et al., 2013 — the basic model is pictured above), I was overcome with the urge to replace it by a simpler system of differential equations.
In the case of chronic myeloid leukemia, the most exciting — and least understood — are the dynamics of stem cells in the bone marrow. The bone marrow stem cells live in a structured cell matrix and this niche is important for protecting them from the more drastic effects that imatinib has in the peripheral blood. In some other cancers — such as the metastasis of prostate cancer to the bone (Araujo et al., under review) — it can be useful to explicitly consider the bone’s internal structure, but for leukemia the spatial structure is a distraction and it is more productive to just follow the existing models and count the cells as shielded without further elaboration. However, spatial structure is not the only possible wrench in the works of an ODE model; the stochastic effects of a low number of discrete cells can have a drastic effect on dynamics (Shnerb et al., 2000). Since we are studying the management of patients with below detection-threshold values of PCR in the blood, it is not unreasonable to believe that the remaining number of cancerous cells in the marrow is also very low. In fact, studies like Lenaerts et al. (2010) rely on the importance of stochastic dynamics for eliminating CML through neutral drift. I was particularly worried about the importance of stochasticity after seeing the relapse threshold of just 200 stem cells in Horn et al. (2013). However, they are actually using in silico populations that are 10 times smaller than their estimates of in vivo counts, and a threshold of 2000 cells (so a thermal variance of just 2%) does not seem as destructive to an ODE model.
The Roeder et al. (2002; 2006; also used in Horn et al., 2013) computational model of CML has three important properties defining each stem cell: its state (quiescent or active), its affinity for activity (or stemness), and its age in the reproductive cycle. While a stem cell is quiescent, it cannot reproduce, but it also hard to target by imatinib or any other treatment; during this time it also regains its affinity for the activity. When a stem cell is active, it gradually loses its affinity for activity, but also proceeds in its cycle towards reproduction. If an active stem cell drops below a certain critical threshold of affinity before reproducing or becoming quiescent again then it will differentiate and leave the bone marrow to become part of the peripheral blood. The most important modeling decision in this system is the mechanism by which cells go from quiescent to active and back. The authors model this as a probability proportional to the product of the affinity of the cell and the sigmoid of the total number of active or quiescent cells; this is suppose to mimic a quorum sensing mechanism that regulates the stem cells from over-reproducing.
 In an ODE model, we can capture this with five compartments for each cell kind (healthy and cancerous, although later we could distinguish between different kinds of cancerous as well). The part we can measure is the ratios of healthy (
In an ODE model, we can capture this with five compartments for each cell kind (healthy and cancerous, although later we could distinguish between different kinds of cancerous as well). The part we can measure is the ratios of healthy (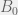
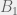


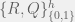
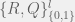
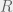
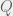
We assume that the initial chromosomal translocation causing the first cancerous stem cells is extremely rare, and so external to our model. For our model, both cell kinds are initially present, and the difference between healthy and non-healthy cells is in the rates that govern their state transitions. In particular, it is believed that the cancerous stem cells turn off their quorum sensing mechanism for transitioning from quiescent to active, and thus are active much more often. Being in the active state more often, means that even with the same reproductive and differentiation rate, the stem cells are more likely to reproduce and differentiate into the peripheral blood. Unfortunately, the peripheral blood is where imatinib has the largest effect (red arrow in the diagram), so it tends to destroy what is being measured very quickly, while affecting the cancerous stem cells much more slowly. However, it is believed to also inhibit the transition from quiescent to active in the cancerous stem cells, allowing the healthy cells to outcompete them on a less immediate timescale. Our goal is to figure out this timescale for each patient, and use that knowledge to guide when it is safe to stop imatinib treatment without relapse.
This model can be obviously extended from 5 to 2m + 1 dimensions per cell kind by increasing the resolution of stem-ness. For instance, if we want m = 3 than we could consider high, medium, and low stem-ness instead of just the current high and low. Since the rates are set independently of the resolution of the stem-ness compartments, the number of parameters stay the same, and the complexity of solving these systems numerically doesn’t increase that much with m. It might seem like reducing the resolution to the coarsest graining of m = 1 would destroy the cyclic nature of the stem-cell activity cycle — that is why I first proposed the m = 2 model as the simplest — but the cyclic dynamics are actually a consequence of the quorum sensing, and the different stem-ness compartments are necessary only to produce different effective reproductive rates of the stem cells depending on how long they spend in active and quiescent states.
Thus, the simplest model has just 3 compartments per cell type. The final major modeling decision is to look at the quorum sensing mechanism. Using the sigmoid as the 



Most importantly, working with a familiar type of system allows us to understand how certain standard separation of scale and homeostasis assumptions will affect our model and use that to analytically solve special cases. The simplification that spring to mind is to notice that although the cancerous stem cells produce a large amount of cancer cells in the peripheral blood, they don’t seem to grow unbounded in the bone marrow. Thus, it is reasonable to assume that the differentiation and reproduction rate of the stem cells (just like the rate of the healthy cells) is at homeostasis; in the m = 1 model it means that the reproduction and differentiation rate are the same (for higher m it is a more complicated question). Also, in the patients we are concentrating on, CML is usually caught during route blood tests well before any noticeable symptoms are manifest. This suggests that although the number of cancerous stem cells might be large, it is still much much smaller than the number of healthy ones. This allows us to make a separation of scales, and assume (for this simplest model) that the effect of the healthy cells is a static offset to the the dynamics of the cancerous cells, and the effect of the cancerous cells is negligible on the dynamics of the healthy cells. Of course, this assumption is not reasonable in the long term, especially for untreated patients, but we only need it for fitting purposes in the relative short-term for patients that are being treated. The assumption allows us to decouple the dynamics of healthy and cancerous cells, and focus on the latter. The resulting system is analytically solvable, but the solution for 
For the last simplification, we can use the fact that the drugs and disease affects are much more pronounced in the peripheral blood than the bone marrow. Hence, we can separate the time-scales of the blood response, and just assume that it equilibrates faster than changes in the marrow occur. This means that 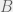

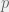
The simple solution (even when it is buried under so many assumptions) allows us to work directly with patient peripheral blood measurements, fit a sigmoid to that time series, and use the parameters of that sigmoid to infer set the parameters of the m = 1 model. Since a sigmoid only has 4 parameters, this means that we could theoretically argue that our model is predictive for any patient who has 5 or more measurements of their peripheral blood. This means we match the minimalist data requirements that Horn et al. (2013) achieved (they fitted two lines with a break), but do so with a completely mechanistic model. Since the m >> 1 shares its basic dynamics with the m = 1 model, we can use the patient parameters plus ones from the literature to instantiate the richer m >> 1 model. This richer model can be simulated numerically without the simplifying assumptions to act as an insilico patient for the doctor to use in considering if treatment can be withdrawn without relapse. Of course, this last paragraph requires a more careful explanation, but you will have to wait for the next blog post for that.
This is my second post ([1]) in a series on mathematical modeling of chronic myeloid leukemia based on the Moffitt Integrated Mathematical Oncology Workshop that I attended this November. The schematic figure of the m = 2 model was made by my team mate Karly Jacobsen, and the whole team contributed essential insights and skills to this project.
References
Araujo, A., Cook, L.M., Lynch, C.C., and Basanta, D. (under review). An integrated computational model of the metastatic prostate cancer-bone microenvironment. Cancer Research.
Horn, M., Glauche, I., Müller, M.C., Hehlmann, R., Hochhaus, A., Loeffler, M., & Roeder, I. (2013). Model-based decision rules reduce the risk of molecular relapse after cessation of tyrosine kinase inhibitor therapy in chronic myeloid leukemia. Blood, 121 (2), 378-84 PMID: 23175686
Lenaerts, T., Pacheco, J. M., Traulsen, A., & Dingli, D. (2010). Tyrosine kinase inhibitor therapy can cure chronic myeloid leukemia without hitting leukemic stem cells. Haematologica, 95(6), 900-907.
Roeder, I., & Loeffler, M. (2002). A novel dynamic model of hematopoietic stem cell organization based on the concept of within-tissue plasticity. Experimental Hematology, 30(8): 853-861.
Roeder, I., Horn, M., Glauche, I., Hochhaus, A., Mueller, M. C., & Loeffler, M. (2006). Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights and clinical implications. Nature Medicine, 12(10): 1181-1184.
Shnerb NM, Louzoun Y, Bettelheim E, & Solomon S (2000). The importance of being discrete: Life always wins on the surface. Proceedings of the National Academy of Sciences of the United States of America, 97 (19): 10322-4.


Pingback: Three goals for computational models | Theory, Evolution, and Games Group
Pingback: Cataloging a year of blogging: applications of evolutionary game theory | Theory, Evolution, and Games Group
Pingback: Misleading models in mathematical oncology | Theory, Evolution, and Games Group
Pingback: Stem cells, branching processes and stochasticity in cancer | Theory, Evolution, and Games Group