Quick introduction: Generalizing the NK-model of fitness landscapes
February 23, 2019 5 Comments
As regular readers of TheEGG know, I’ve been interested in fitness landscapes for many years. At their most basic, a fitness landscape is an almost unworkably vague idea: it is just a mapping from some description of organisms (usually a string corresponding to a genotype or phenotype) to fitness, alongside some notion of locality — i.e. some descriptions being closer to each other than to some other descriptions. Usually, fitness landscapes are studied over combinatorially large genotypic spaces on many loci, with locality coming form something like point mutations at each locus. These spaces are exponentially large in the number of loci. As such, no matter how rapidly next-generation sequencing and fitness assays expand, we will not be able to treat a fitness landscape as simply an array of numbers and measure each fitness. At least for any moderate or larger number of genes.
The space is just too big.
As such, we can’t consider an arbitrary mapping from genotypes to fitness. Instead, we need to consider compact representations.
Ever since Julian Z. Xue first introduced me to it, my favorite compact representation has probably been the NK-model of fitness landscapes. In this post, I will rehearse the definition of what I’d call the classic NK-model. But I’ll then consider how the model would have been defined if it was originally proposed by a mathematician or computer scientists. I’ll call this the generalized NK-model and argue that it isn’t only mathematically more natural but also biologically more sensible.
For simplicity of presentation, let’s focus on biallelic systems.
The classic NK-model is a fitness landscape on 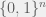
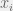

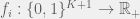
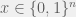
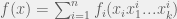
By varying K we can control the amount of epistasis in the landscape. With K = 0 we have a smooth landscape, and for higher K we can get various kinds of epistasis. The model also provides an upper bound of 
Consider the simplest non-trivial case: K = 1. Here, the fitness function will only have at most n fitness components, with each involving two loci. That means a connected gene-interaction graph would be either a tree or a tree with an extra edge. And a disconnected graph would have each component as a tree or tree with an extra edge. This seems incredibly restrictive given the 
I think that a computer scientists or mathematician would never have given such a definition an awkward definition. So let me propose another one (again, restricting to biallelic systems for simplicity of presentation).
The generalized NK-model is a fitness landscape on 
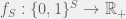
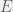
![x[S]](https://s0.wp.com/latex.php?latex=x%5BS%5D&bg=f0f0f0&fg=555555&s=0&c=20201002)
![f(x) = \sum_{S \in E} f_S(x[S])](https://s0.wp.com/latex.php?latex=f%28x%29+%3D+%5Csum_%7BS+%5Cin+E%7D+f_S%28x%5BS%5D%29&bg=f0f0f0&fg=555555&s=0&c=20201002)
Now, if we turn back to the K = 1 case, we can have all possible
More importantly, this allows our definition of the NK-model to more clearly align with well studied theoretical computer science topics like valued-constraint satisfaction problems. For the details of the correspondence, take a look Alexandru Strimbu’s post. This correspondence is probably the biggest mathematical advantage of the generalized definition.
But the advantage isn’t limited to mathematics. I think there is also an important aspect of biological interpretation to consider.
In particular, the classic NK-model seems to enshrine — or at least heavily favour — the one-gene one-function view of molecular biology.
The easiest way to interpret the fitness components in the NK-model is as ‘basic functions’ that together add up to the total fitness of the organism. In this way, the fitness components serve as a rudimentary decomposition of the genotype->phenotype map. But in this interpretation, if each gene is linked to a single fitness component then we are linking one gene to one function. Sure, that function is mediated by K other genes, but there are still no more of them than there are genes.
This seems like a strange dogma to just build into our model.
With the generalized model, we can avoid this. Instead we can just talk of each 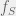
What do you think, dear reader?


A box of useful tools for life in boolean spaces (and species) can be found here —
☞ Differential Logic and Dynamic Systems
It is important to recognize that the concept of adaptive landscapes is very old (1932), and it is one of the first complex systems models. As such the original language, math, and concepts are clumsy and often simplistic. I agree that the models need updating, and your suggestion is a good one. However, I want to admonish people to not fault the early pioneers for getting the details wrong.
Thanks for that reminder Charles!
I didn’t want to imply fault with early pioneers, especially Wright. He was certainly not talking about the NK-model, after all. I am also not aiming to use the model for the goals that he had in mind (and not even the goals that Kauffman & Levin had in mind), so it is not surprising that I’ll need to tweak things for new purposes.
But I do think it is important to update our math to be less clumsy. There is a tendency to grandfather-in models just because they are old. The tweak I propose here is certainly not original, but it does make it easier to interface biology with theoretical computer science (which is my overarching goal).
Pingback: Game landscapes: from fitness scalars to fitness functions | Theory, Evolution, and Games Group
Pingback: The gene-interaction networks of easy fitness landscapes | Theory, Evolution, and Games Group