Ratcheting and the Gillespie algorithm for dark selection
November 14, 2017 3 Comments
In Artem’s previous post about the IMO workshop he suggests that “[s]ince we are forced to move from the genetic to the epigenetic level of description, it becomes important to suggest a plausible mechanism for heritable epigenetic effects. We need to find a stochastic ratcheted phenotypic switch among the pathways of the CMML cells.” Here I’ll go into more detail about modeling this ratcheting and how to go about identifying the mechanism. We can think of this as a potential implementation of the TYK bypass in the JAK-STAT pathway described experimentally by Koppikar et al. (2012). However, I won’t go into the specifics of exact molecules, keeping to the abstract essence.

After David Robert Grime’s post on oxygen use, this is the third entry in our series on dark selection in chronic myelomonocytic leukemia (CMML). We have posted a preprint (Kaznatcheev et al., 2017) on our project to BioRxiv and section 3.1 therein follows this post closely.
To make this model “…as simple as possible, but no simpler” I’ll begin with an ODE and ask what additional features the model needs in order to be effective. The first requirement to see ratcheting is an unstable equilibrium in between the “sensitive” and “resistant” concentrations of a protein complex in the cell. This unstable equilibrium prevents a spontaneous change in all cells that would occur in the time scale of chemical reactions. This, however, means we cannot use a basic ODE to model our chemical interactions. In order for a select few cells to cross this unstable equilibrium and become “resistant” we need a certain degree of stochasticity. This is where the Gillespie algorithm comes in.
Instead of iterating over constant time steps (as you might using Euler’s method to model an ODE) the Gillespie algorithm iterates over events and then assigns a time to the event after the fact. The time (until another event occurs) is assigned scholastically, usually with the memoryless exponential probability distribution function 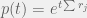
Where t is time, and 

We then add and subtract species as per the event and assign the chosen time to the event.
The rates are then determined by the number of ‘species’, in biology this would be the number of cells of a certain type or the population of a particular animal, in chemistry it can be the number of molecules.
Let’s begin with a simple reaction where one type of chemical transforms into another 
One can work out that a stable equilibrium occurs at 

If we then converted AB to two B’s we would have a classic autocatalysis reaction. But that is not enough for our purposes since this reaction relies just as much on A as it does on B[2]. We therefore need a “peer-pressure” reaction step: 
Here A is outnumbered by B in the complex and is therefore converted to a B. We do the same for B’s converting to A’s: 
Note that the net reaction is still one A being converted to one B. If we give reaction rate coefficients of 1 to all reactions except those where complexes are used up (which will have coefficients that are >>1 in order to keep the concentration of complexes small) we can approximate the change in the number of B molecules as:
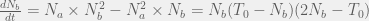
where 

One last thing. If we plug these values into the Gillespie algorithm the probability of having an A turn into a B when there are no other B’s around is 0. Therefore, in order to have a stochastic change in state, we need a leak equation where the net reaction (
These reactions act as both ‘mutation’ and mode of inheritance. Stochastic changes in the amount of chemical due to the ‘leak’ reactions can lead to a spontaneous change in state in some cells, mutation (as seen in picture 2 but not picture 1). But other cells can also impose this state by increasing the number of molecules of one chemical species, either through diffusion or through activation by some other diffusing species: inheritance. Hence two requirements, variation and inheritance, can be satisfied by a relatively simple set of chemical reactions. If the “spreader” of B is more concentrated and/or can spread further in the presence of a drug then proliferation of this state will increase. Leading to the third and final requirement for Darwinian evolution: selection.
Notes and References
- When programming a stochastic model it is usually necessary to convert a random variable from the uniform distribution to one in our desired probability distribution, this is how it can be done for exponential distributions:
- We need the reaction to rely more on B since unstable equilibria rely on the predominance of the species being created, this is also why we need autocatalysis.

Kaznatcheev, A., Grimes, D. R., Vander Velde, R., Cannataro, V. L., Baratchart, E., Dhawan, A., … & Basanta, D. (2017). Dark selection for JAK/STAT-inhibitor resistance in chronic myelomonocytic leukemia. BioRxiv, 211151.
Koppikar, P., Bhagwat, N., Kilpivaara, O., Manshouri, T., Adli, M., Hricik, T., … & Leung, L. (2012). Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature, 489(7414): 155-159.


Pingback: Dark selection from spatial cytokine signaling networks | Theory, Evolution, and Games Group
Pingback: Cataloging a sparse year of blogging: IMO workshop and preprints | Theory, Evolution, and Games Group
Pingback: Blogging community of computational and mathematical oncologists | Theory, Evolution, and Games Group