Programming playground: A whole-cell computational model
July 23, 2012 13 Comments
Three days ago, Jonathan R. Karr, Jayodita C. Sanghvi and coauthors in Markus W. Covert’s lab published a whole-cell computational model of the life cycle of the human pathogen Mycoplasma genitalium. This is the first model of its kind: they track all biological processes such as DNA replication, RNA transcription and regulation, protein synthesis, metabolism and cell division at the molecular level. To achieve this, the authors integrate 28 different sub-models of the known cellular processes.

Figure 1A from Karr, Sanghvi et. al (2012): A diagram of the 28 sub-models, colored by category: RNA (green), protein (blue), metabolic (orange), DNA (red). The modules are connected by arrow representing common metabolites (orange), RNA (green), proteins (blue), and DNA (red).
The key technical accomplishment was integrating the 28 modulus into a single model. Each module is based on existing models, but different modules are expressed in different paradigms: ODE, Boolean, probabilistic, and constraint-based. For me, this is the most impressive aspect of the work. Usually, when I look at biology (or psychology), I see a mishmash of models with each expressed in its own language and seemingly incompatible with the others. The authors overcame this by assuming the modulus are independent on short timescales (under 1 second). This allows the software to keep track of 16 global cell variables which are used as inputs for the submodulus that are run to simulate 1 second and their results used to update the global variables and repeat the loop. The whole software is available online and the authors can use the data gathered to produce a video of a single cell’s life cycle:
The authors show that the model has a high level of agreement with existing data. They also use the predictions to run several novel real-biology experiments, and even partially overturn (or complete) a previous experimental observation based on hints from their model. In particular they show that disruption of the IpdA gene — which Glass et al. (2006) suggested as non-essential — has severe (but noncritical) impact on cell growth. I wish I could comment more on the validity of the model as judged by experiments, but molecular biology is magic to me.
The simulation results that were most exciting for me was looking at the effects of single-gene disruptions on phenotype. The bacterium Mycoplasma genitalium is a human urogenital parasite whose genome contains 525 genes (Fraser et al., 1995). It is not an easy model organism to work with, but it has the smallest known genome that can constitute a cell. Part of the team on this project, is from J. Craig Venter Institute and has extensive experience with the organism due to their effort to create the first self-replication synthetic life by implanting artificial DNA into Mycoplasma genitalium. I would not be surprised if this model plays a vital part in the institute’s engineering.
Karr, Sanghvi et al. (2012) ran simulations of each of the 525 possible single-gene disruption strains. They found that 284 genes were essential to sustain growth and division and 117 are non-essential — a 79% agreement with the experimental results of Glass et al. (2006). Of particular interest for me was that in some cases it took more than one generation for specific proteins to fall to lethal levels. As far as I understand this is because when a single-cell divides, daughters get both a copy of the mother DNA and have their initial levels of proteins and RNA set to within statistical fluctuations of those of their mother. Due to my complete lack of basic biological background, this seemed an interesting example of Lamarkian evolution. In particular, it raises questions on how to best combine single-cell learning and evolution. From a naive Bayesian model of learning, it would seem that this would allow cells to pass on their priors — a biological evolution counterpart to Beppu & Griffiths (2009) cultural ratchet.
The detail of the whole-cell model is impressive. I hope that the software becomes a tool for theorists without access to a wet-lab to play around with cells. The approach is an antithesis to the simple and completely unrealistic models I am accustomed to building. For me, it raises many thoughts on how to better think about the distinction between genotype and phenotype that is almost always ignored in evolutionary game theory. For now the whole-cell model is computationally too expensive for me to build evolutionary dynamics from it, but maybe parts of the code can be simplified or ignored or maybe we could use more course-grained models. Either way, I am excited for my new playground!
References
Beppu, A., & Griffiths, T. (2009). Iterated learning and the cultural ratchet. Proceedings of the 31st Annual Conference of the Cognitive Science Society, 2089-2094.
Fraser, C.M., Gocayne, J.D., White, O., Adams, M.D., Clayton, R.A., Fleischmann, R.D., Bult, C.J., Kerlavage, A.R., Sutton, G., Kelley, J.M., et al. (1995). The minimal gene complement of Mycoplasma genitalium. Science 270, 397–403.
Glass, J.I., Assad-Garcia, N., Alperovich, N., Yooseph, S., Lewis, M.R., Maruf, M., Hutchison, C.A., Smith, H.O., & Venter, J.C. (2006). Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. USA 103, 425–430.
Karr, J.R., Sanghvi, J.C., Macklin, D.N., Gutschow, M.V., Jacobs, J.M., Bolival, B., Assad-Garcia, N., Glass, J.I., & Covert, M.W. (2012). A whole-cell computational model predicts phenotype from genotype Cell, 150, 389-401 DOI: 10.1016/j.cell.2012.05.044
 -bit program that takes the current organism A and outputs a new organism B of higher fitness (note, that it needs an oracle call for the Halting-problem to do so). The probability that A is replaced by B is then
-bit program that takes the current organism A and outputs a new organism B of higher fitness (note, that it needs an oracle call for the Halting-problem to do so). The probability that A is replaced by B is then  . There is no way to decouple the selection step from the mutation step in an algorithmic mutation, although this is not clear without the technical details which I will postpone until a future post. Chaitin’s model does not have random mutations, it has randomized directed mutations.
. There is no way to decouple the selection step from the mutation step in an algorithmic mutation, although this is not clear without the technical details which I will postpone until a future post. Chaitin’s model does not have random mutations, it has randomized directed mutations.
 is a halting self-delimiting program, and
is a halting self-delimiting program, and 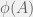 is the integer
is the integer  to mean the running time of
to mean the running time of 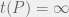 for a non-halting program
for a non-halting program 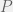 .
.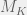 works as follows.
works as follows.  calculates a lowerbound
calculates a lowerbound 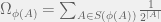 of
of 
 is the set of programs smaller than
is the set of programs smaller than 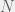 bits in length and halting by
bits in length and halting by  and mutates
and mutates  . Here
. Here  is a self-delimiting prefix that finds the first
is a self-delimiting prefix that finds the first  , outputs
, outputs  then
then 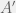 a halts and is thus an organism with fitness
a halts and is thus an organism with fitness 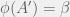 . If
. If 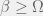 then
then 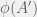 ). Chaitin’s mutations either return a strictly more fit organism, or do not return one at all (equivalently: an organism with undefined fitness).
). Chaitin’s mutations either return a strictly more fit organism, or do not return one at all (equivalently: an organism with undefined fitness).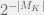 ). This is probably why he writes them as ‘random’, however picking a directed mutation at random, is just randomized directed mutation. The concept of a mutant having lower fitness, is undefined in metabiology. A defense would be to say that algorithmic mutations actually combine the steps of mutation and selection into one — this is a technique that is often used in macroevolutionary models. However, I cannot grant this defense, because in macroevolutionary models, even when a fitter agent is always selected, the mutations are still capable of defining an unfit individual. Further, when macroevolutionary models set up such a hard gradient (of always selecting the fitter mutant) then these models are not used to study fitness because it is trivial to show unbounded fitness growth in such models.
). This is probably why he writes them as ‘random’, however picking a directed mutation at random, is just randomized directed mutation. The concept of a mutant having lower fitness, is undefined in metabiology. A defense would be to say that algorithmic mutations actually combine the steps of mutation and selection into one — this is a technique that is often used in macroevolutionary models. However, I cannot grant this defense, because in macroevolutionary models, even when a fitter agent is always selected, the mutations are still capable of defining an unfit individual. Further, when macroevolutionary models set up such a hard gradient (of always selecting the fitter mutant) then these models are not used to study fitness because it is trivial to show unbounded fitness growth in such models. where
where 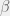 is some rational number greater than 0 and less than
is some rational number greater than 0 and less than  do N = N + 1 end
do N = N + 1 end but we will see that it is a superficial feature.
but we will see that it is a superficial feature. 

 (in fact, the number doesn’t even need to be irrational it just has to have a non-terminating expansion in base 2). In other words, the algorithmic part of metabiology is superficial.
(in fact, the number doesn’t even need to be irrational it just has to have a non-terminating expansion in base 2). In other words, the algorithmic part of metabiology is superficial.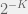 and – or + uniformly. Now, we can define a random mutation operator:
and – or + uniformly. Now, we can define a random mutation operator:

 we can use our favorite selection rule to chose who survives. A tempting one is to use Chaitin’s hard-max and let whoever is larger survive. Alternatively, we can use a softer rule like
we can use our favorite selection rule to chose who survives. A tempting one is to use Chaitin’s hard-max and let whoever is larger survive. Alternatively, we can use a softer rule like  (this is a popular rule in evolutionary game theory models). Alternatively, we can stick ourselves plainly inside
(this is a popular rule in evolutionary game theory models). Alternatively, we can stick ourselves plainly inside  possible tags, and interactions happen only within a tag. The agent can be either a cooperator (ethnocentric in our terminology) or defector (selfish in our terminology), allowing for a total of
possible tags, and interactions happen only within a tag. The agent can be either a cooperator (ethnocentric in our terminology) or defector (selfish in our terminology), allowing for a total of 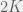 phenotypes. The payoffs from random interactions are computed according to the standard b-c Prisoner’s dilemma game, with
phenotypes. The payoffs from random interactions are computed according to the standard b-c Prisoner’s dilemma game, with  the benefit of receiving cooperation and
the benefit of receiving cooperation and  the cost of providing the cooperation. The population is updated by selection two individuals uniformly at random and having the first adopt the strategy of the second (with some probability of mutation) with probability
the cost of providing the cooperation. The population is updated by selection two individuals uniformly at random and having the first adopt the strategy of the second (with some probability of mutation) with probability 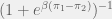 where
where 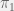 is the utility of the first individual, and
is the utility of the first individual, and  — the second. The parameter
— the second. The parameter  ) is considered.
) is considered. , and the probability of mutating both strategy and tag at the same time is given by
, and the probability of mutating both strategy and tag at the same time is given by  . With this Traulsen & Nowak arrive at their main result, cooperative behavior evolves if:
. With this Traulsen & Nowak arrive at their main result, cooperative behavior evolves if:
 where the first bit gives the strategy and the remaining
where the first bit gives the strategy and the remaining  bits are the tag. They assume that any bit has a mutation probability
bits are the tag. They assume that any bit has a mutation probability  , but do not allow double mutations in the tag. This gives the condition for cooperation:
, but do not allow double mutations in the tag. This gives the condition for cooperation:
 . However, it is not clear to me why the authors didn’t simply allow an arbitrary number of mutations in the tag-coding region. This would give
. However, it is not clear to me why the authors didn’t simply allow an arbitrary number of mutations in the tag-coding region. This would give  and
and 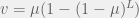 which provides a simpler condition of:
which provides a simpler condition of:
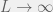 ) and small (relative to size of genome) mutations
) and small (relative to size of genome) mutations  gives:
gives:
 is greater than
is greater than  .
. ) they arrive at the requirement for cooperation:
) they arrive at the requirement for cooperation: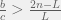
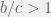 for linked strategy-tag genes, thus recreating (in-spirit) the earlier results of Riolo et al. (2001) and Traulsen & Schuster (2003).
for linked strategy-tag genes, thus recreating (in-spirit) the earlier results of Riolo et al. (2001) and Traulsen & Schuster (2003).
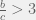 in the minimal case of 2 tags, and the requirements relax towards the trivial
in the minimal case of 2 tags, and the requirements relax towards the trivial  as
as  . In my
. In my  which is higher than the 5/3 required from Traulsen & Nowak’s results, and observed both tides of tolerance (periodic fluctuations in amount of cooperative interactions) and absorbing states of all-cooperation or all-defection. I also briefly showed that
which is higher than the 5/3 required from Traulsen & Nowak’s results, and observed both tides of tolerance (periodic fluctuations in amount of cooperative interactions) and absorbing states of all-cooperation or all-defection. I also briefly showed that  bifurcates, and this can be even more clearly seen in the following graph of
bifurcates, and this can be even more clearly seen in the following graph of  with only two tags:
with only two tags:

